TF-MINDI: Transcription Factor Motif Instance Neighborhood Decomposition and Interpretation#


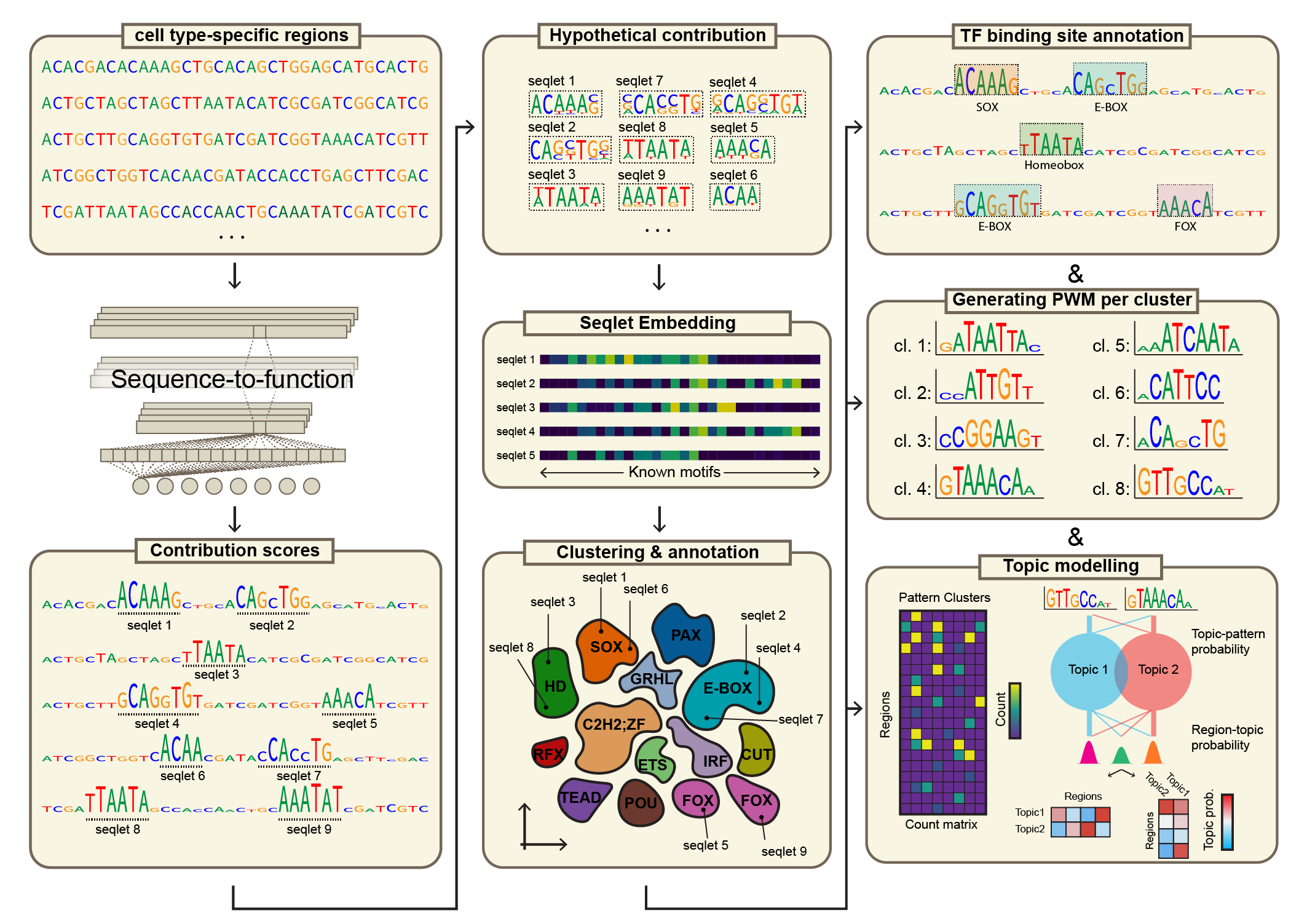
TF-MINDI is a Python package for analyzing transcription factor binding patterns from deep learning model attribution scores. It identifies and clusters sequence motifs from contribution scores, maps them to DNA-binding domains, and provides comprehensive visualization tools for regulatory genomics analysis.
Getting Started#
Please refer to the documentation for detailed tutorials and examples, in particular, the API documentation and Tutorials
Key Features#
Seqlet Extraction: Identifies important sequence regions from contribution scores using recursive seqlet calling from
tangermemeMotif Similarity Analysis: Compares extracted seqlets to known motif databases using TomTom
Clustering & Dimensionality Reduction: Groups similar seqlets using Leiden clustering and t-SNE visualization
DNA-Binding Domain Annotation: Maps seqlet clusters to transcription factor families
Pattern Generation: Creates consensus motifs from clustered seqlets with alignment
Comprehensive Visualization: Region-level contribution plots, t-SNE embeddings, motif logos, and heatmaps
Installation#
tfmindi is compatible with python version 3.10-3.12 and requires a high performance computing (HPC) environment with Linux as operating system.
CPU Version (Default)#
pip install tfmindi
GPU-Accelerated Version (Recommended for large datasets)#
# Requires CUDA-compatible GPU (CUDA 12.X)
pip install tfmindi[gpu]
The GPU version provides significant speedups for:
PCA computation
Neighborhood graph construction
t-SNE embedding
Leiden clustering
We’re still working on making the tfmindi package as GPU-compatible as possible.
If tfmindi can’t find the GPU, try importing rapids_singlecell directly in python and see what errors you get.
You might have to explicitly set your LD_LIBRARY_PATH for cuml as described here.
Quick Start#
TF-MINDI follows a scanpy-inspired workflow:
Preprocessing (
tm.pp): Extract seqlets, calculate motif similarities, and create an Anndata objectTools (
tm.tl): Cluster seqlets and create consensus patternsPlotting (
tm.pl): Visualize results
import tfmindi as tm
# Optional: Check GPU availability and set backend
print(f"GPU available: {tm.is_gpu_available()}")
print(f"Current backend: {tm.get_backend()}")
# tm.set_backend('gpu') # Force GPU backend
# tm.set_backend('cpu') # Swap back to CPU backend
# Extract seqlets from contribution scores
seqlets_df, seqlet_matrices = tm.pp.extract_seqlets(
contrib=contrib_scores, # (n_examples, 4, length)
oh=one_hot_sequences, # (n_examples, 4, length)
threshold=0.05
)
# Calculate motif similarity
motif_collection = tm.load_motif_collection(
tm.fetch_motif_collection()
)
similarity_matrix = tm.pp.calculate_motif_similarity(
seqlet_matrices,
motif_collection,
chunk_size=10000
)
# Create AnnData object for analysis
adata = tm.pp.create_seqlet_adata(
similarity_matrix,
seqlets_df,
seqlet_matrices=seqlet_matrices,
oh_sequences=one_hot_sequences,
contrib_scores=contrib_scores,
motif_collection=motif_collection
)
# Cluster seqlets and annotate with DNA-binding domains
tm.tl.cluster_seqlets(adata, resolution=3.0)
# Generate consensus logos for each cluster
patterns = tm.tl.create_patterns(adata)
# Visualize results
tm.pl.tsne(adata, color_by="cluster_dbd")
tm.pl.region_contributions(adata, example_idx=0)
tm.pl.dbd_heatmap(adata)
Release Notes#
See the changelog.
Contact#
If you found a bug, please use the issue tracker.